FDA(美国食品和药物管理局Food and Drug Administration)对医疗器械的管理通过器械与放射健康中心(CDRH)进行的,中心监督医疗器械的生产、包装、经销商遵守法律下进行经营活动。

讲医疗器械FDA之前,我们先讲FDA分类,以及了解PMA,HDE,De Novo,IDE是什么?
医疗器械FDA如何分类?
美国FDA医疗器械产品目录中共有1700多种。根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ)。风险等级逐级升高,Ⅲ类风险等级最高。
一般控制 绝大部分产品只需进行注册、列名和实施GMP规范(良好的生产规范),产品即可进入美国市场(其Ⅰ类产品(占47%左右)
中极少数产品连GMP也豁免,极少数保留产品则需向FDA递交510(K)(产品上市登记)申请即PMN(上市前的通告)) 这些器材只要经过一般控制就可以确保其功效与安全性,如拐杖、眼镜片、胶布等,约占全部医疗器材的27%。 这些控制包括: 禁止粗制滥造及不当标示的产品销售; FDA得禁止不合格产品销售; 必须报告FDA有关危害性、修理、置换等事项; 限制某些器材的贩卖、销售、及使用; 实施GMP(Good Manufacturing Practice)。
一般控制 + 特殊控制 企业在进行注册和列名后,这些产品除了上述一般控制之外,其余大多数产品均要求进行上市前通告(PMN:Premarket Notification)(即510K)。少量的II类产品可以豁免上市前通告程序。生产企业须在产品上市前90天向FDA提出申请,通过510K审查后,产品才能够上市销售。
一般控制 + 上市前许可(Premarket Approval) 企业在进行注册和列名后,须实施GMP并向FDA递交PMA(产品上市审核标准)申请(部分Ⅲ类产品还是PMN)。 一般来说, III类产品多为维持、支持生命或植入体内的器材,对病患具有潜在危险,可能引起伤害或疾病者,如心律调节器、子宫内器材及婴儿保温箱等,约占所有器材的8%。这些器材必须取得FDA的PMA之后方能销售。
PMA,HDE,De Novo,IDE是什么?
什么是PMA?
PMA是FDA监管最严格的III类医疗器械的上市前提交类型,申请PMA,也就是III类医疗器械的注册申请,必须充分证明其安全性和有效性,通过IDE申请临床试验,提交大量的研发文档和技术文档。
什么是HDE?
人道主义器械豁免可用于疾病或者少于每年8000人的情况—如罕见的右心衰竭。他们可以在没有临床证据的情况下得到豁免,申请HUD再由HDE审批,但是申请人必须对产品安全性作出解释并使人相信其应用利大于弊。
什么是De Novo?
De Novo(产品风险等级的重新分类)是一种基于风险的分类过程。对于没有合法上市对比产品的新型医疗器械,即使是中低风险,仍无法通过510(k)申请建立实质等同(SE)从而获得上市许可。针对这类产品,FDA建立了De Novo申请途径。
De Novo使得更多符合现代性能的新型器械上市,并可以作为510 (k)申请路径实质等同评价的对比产品。同时,FDA也会采取新方法,促进在510(k)申请路径的实质等同对比过程中使用更加现代的对比产品,从而又促进更多的医疗器械采用De Novo途径。
什么是IDE?
IDE指的是试验用器械豁免,是FD&C Act对于仅用于临床试验的医疗器械的管制措施,其主要的精神是让研究发展中的医疗器械可以免除掉对于以上市销售为目的的器械产品的种种管制,而以较简单的方式让制造商通过临床试验来收集安全性和有效性的信息资料,从而为510(k)和PMA申请提供数据支撑。FD&C Act 520(g)条款中授权FDA对临床试验用的器械,可以免除企业注册、产品登记、标示、510(k)、PMA或医疗器械伤害报告等规定,除设计控制外,也不必遵循质量体系法规的要求,但仍要具有与上市时同样的合格标准(也就是说,制造商必须确保临床试验用的医疗器械已经符合安全性评估),遵守特别的标识规定(如注明限临床试验使用或警告语),制造商也不准对临床试验用的医疗器械有广告、商业化或任意延长临床试验期限的行为。对于大多数的PMA申请而言,临床试验要求是必须的。然而,制造商在提交510(k)申请的时候,却仅在少数情况下要求提供临床试验数据。这里“试验”还包括对已合法上市器械的改进和新用途的临床评价。除豁免的情况外,所有试验用器械在其临床试验启动前都必须取得IDE许可。
什么是US Agent美国代理人?
美国代理人是指住在美国或者在美国有经营地点。美国代理人不可以用邮政信箱作为地址。美国代理人不能使用只是一个接听服务。他们必须能够在正常的工作时间有相关的员工接听电话。
美国代理人的职责包括:
-协助FDA和国外工厂沟通
-回答关于国外工厂的器械进口到美国的一些问题
-协助FDA安排国外工厂FDA审查
如果FDA不能够直接或者迅速地联系国外工厂,FDA可以提供信息或者文件给到美国代理人,这个行动实质等同于把信息直接给到国外工厂。请注意美国代理人没有职责上报医疗器械不良事件依据医疗器械报告法规21 CFR Part 803),或者提交510(k)文件 (21 CFR Part 807,SubpartE)
CFR中有关医疗器械的法规:
? 21 Code of Federal Regulations (CFR): Parts 800-1050
– 800-861: cross-cutting device requirements(横切装置要求)
? Example: 812 - Investigational Device Exemption(研究器械豁免)
– 862-1050: device-specific requirements(特定设备要求)
? Example: 876 - Gastroenterology and Urology Devices(肠胃科和泌尿科器械)
? 21 CFR: Parts 1-99
– general medical requirements that also apply to medical devices(适用于医疗器械的一般医疗要求)
医疗器械注册流程:
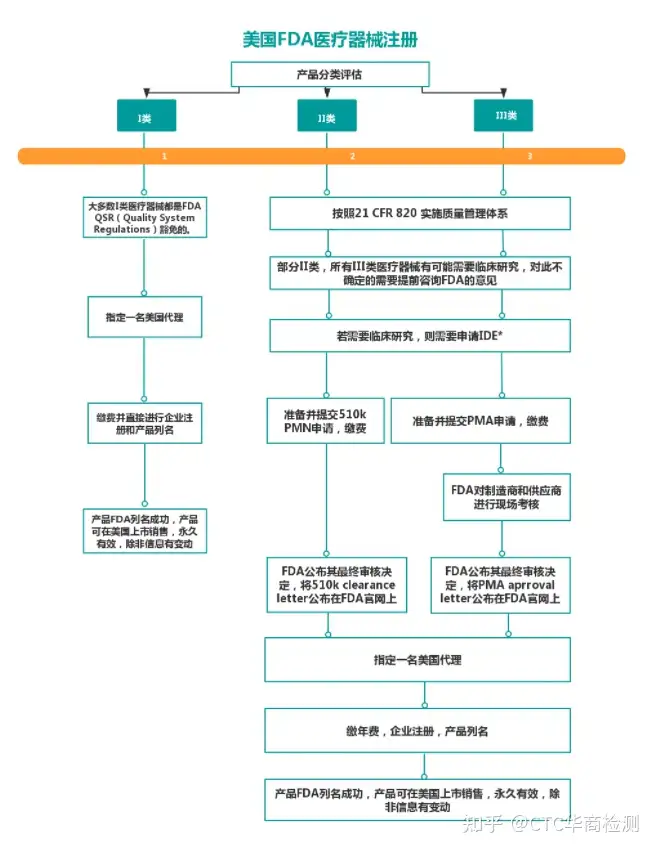
医疗器械FDA验厂:
针对美国市场,其中FDA注册是强制性要求,普通FDA注册和产品列名是针对一般低风险产品,行业内称作510K豁免产品,510K则对应针对高风险产品,也就是我们说的510k产品。
FDA验厂针对部分被抽查到的企业,一般产品风险越高,被抽到验厂的概率越大。医疗器械FDA验厂来说,医疗器械FDA验厂是按照QSR820体系,QSR820是法规,不是认证,厂家拒绝检查视为违法。拒绝后最直接的后果就是会收到FDA的警告信,最严重的后果就是会被拉到黑名单,并被FDA官网出具进口禁令,从此无缘美国市场。
目前FDA对中国制药和器械企业的审核数量约为130-160家/年。对药企而言,注册企业约为800家。而器械则超过3400家,对于I类产品,每四年检查一次,对于II/III类产品,每两年检查一次。
FDA认证常见问题:
问题一: FDA证书是哪个机构发放的?
答:FDA注册是没有证书的,产品通过在FDA进行注册,将取得注册号码,FDA会给申请人一份回函(有FDA行政长官的签字),但不存在FDA证书说法。
问题二: FDA需要指定的认证实验室检测吗?
答:FDA是一个执法机构,而不是服务机构,如果有人说他们是FDA下属的认证实验室,那么他至少是在误导消费者,因为FDA没有面向公众的服务性认证机构与实验室,也没有所谓的“指定实验室”。FDA作为联邦执法机构,不可以从事这种既当裁判又当运动员的事。FDA只会对服务性的检测实验室的GMP质量进行认可,合格的颁发合格证书,但不会向公众“指定”,或推荐特定的一家或几家。
问题三: FDA注册是否一定需要一位美国代理人?
答:是的,中国申请人在进行FDA注册时必须指派一名美国公民(公司/社团)作为其代理人,该名代理人负责进行位于美国的过程服务,是联系FDA与申请人的媒介。


部分过审案例