2017年5月25日体外诊断医疗器械法规(In Vitro Diagnostic Regulation, IVDR)正式生效,并于2022年5月26日起实施。自实施之日起,IVDR将取代原体外诊断设备指令(IVDD)。在欧盟地区范围内,医疗器械制造商必须按照欧盟MDR以及IVDR的规定对产品、文件和生产流程进行调整,才能在欧洲经济区(EEA)发售。

今年10月,欧盟颁布了医疗器械新法规IVDR延期的草案,该草案修正了该过渡期,数量众多的低风险等级器械将会有更长的过渡期,并且,由发证机构介入发证的IVD产品的过渡期将会延长到2025年5月26日。
分别延期调整如下:

对于IVDD产品可以在市场销售的时间,也相应做出了调整如下:

委员会为什么建议延长过渡期?
由于医疗资源被转移用于同时应对新冠肺炎疫情以及IVDR引入的重大变化,成员国当局、医疗机构、公告机构和经济运营商无法在2022 年 5 月 26 日前完全满足IVDR的实施要求。
尤其是迄今为止,根据IVDR仅指定了十家公告机构,公告机构审核资源严重不足,导致制造商难以及时进行法律规定的合格评定程序。因此,对于在其他成员国设立的中小企业(SMEs)来说,面临更大的困难,它们倾向于向自己或邻近成员国的公告机构提出申请。
如果不加以解决,这种情况可能会导致市场上为医疗机构和公众提供的大量体外诊断医疗器械的供应出现严重中断。
IVDR第110条规定,在2022年5月26日之前,持有公告机构根据IVDD颁发的证书的体外诊断医疗器械的过渡期至2024年5月26日。这一过渡期将延长1年,至2025年5月。然而,只有在该指令下(约8%)获得公告机构证书的医疗器械才会从现有的过渡条款中受益。
从IVDD到IVDR的主要变化是什么?
1.分类要求变化:
更新后的IVDR将所有体外诊断设备从低到高分成A、B、C、D四类,该分类规则来源于全球协调工作组(GHTF)。
2.增加公告机构(NB)的介入
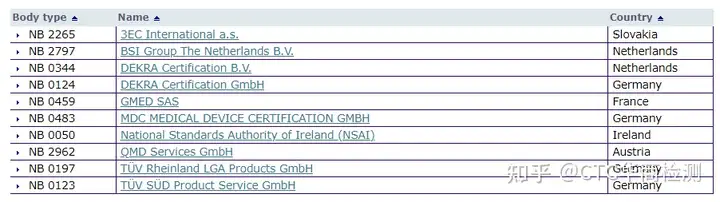
最主要变化点在于公告机构(Notified Body,NB)介入的增加,涉及产品包括部分A类和所有的B、C、D类。在整个IVD领域,涉及公告机构介入的产品数量从IVDD监管体系下的10%~20%增加至80%~90%。以下是授权NB公告号详细信息。目前已经增至10家。
链接:https://ec.europa.eu/growth/too
3.医疗器械数据库(Eudamed)的建立
在IVDR的法规体系下,欧盟主管当局一个很重要的工作就是未来将要推出医疗器械数据库(Eudamed),该数据库涵盖从产品上市前审批到上市后监管中的很多重要信息。
4.医疗器械唯一标识系统(UDI)的引入
IVDR中所提出的UDI由一个固定的产品识别码(Device Identifier,DI)和一个非固定的生产识别码(Production Identifier,PI)组成。生产企业在实施UDI的过程中,需要明确各产品的UDI代码及所包含的信息,并在产品上加贴UDI标贴,同时以电子形式存储UDI相关信息并在Eudamed系统上申报,用以增强产品的追溯以及上市后的管理。
5.新增监管负责人
IVDR法规中,首次要求每个制造商企业内,至少有一位法规负责人,负责处理与产品相关的监管、合规性相关工作,作为医疗器械公司和指定机构之间的联系。
体外诊断试剂的认证主要包括5种不同的方式:
1) Annex ll EC符合性自我声明;
2) Annex lV EC符合性声明(全面质量保证体系);
3) Annex VEC型式检查;
4) Annex VIEC产品验证;
5) Annex VII EC符合性声明(生产质量保证体系)。
另:分类属于“Other”的产品在满足IVDD指令要求后,通过Anne lI途径,在产品上使用CE标志,无需公告机构参与。
认证流程:
1) 按照IVDD 2017/746编制技术文件;
2) 指定欧盟授权代表;
3) 完成IVDD 2017/746器械注册;
4) 发布DOC文件;
5) 按照IVDD 2017/746执行上市后监督体系。
对于IVDD不需要公告机构参与,但是IVDR需要公告机构参与的IVD器械制造商,应尽快(在2022年5月26日之前)按照IVDD来完成DOC以获得缓冲期资格。